Research
概要
生理機能をつかさどるペプチド・タンパク質は、創薬のための魅力的なリード化合物であるととともに、新しい創薬研究の標的分子でもあります。薬品化学分野では、微量で強力な生物活性を示すペプチドをもとにして、医薬品として利用可能な優れた特性を有する化合物の創製研究を進めています。また、最新のタンパク質の化学合成法を活用することにより、新たな医薬シーズを探索するための新技術の開発に取り組んでいます。
薬品化学分野における創薬研究では、主としてがん、感染症、中枢神経疾患を対象として取り扱っています。研究において創製したリガンドや阻害剤は、これらに分類されない疾患に対しても効果を示すことがあることから、標的分子の種類に応じてその応用範囲は多岐にわたります。また、優れた生物活性を示す化合物は、生命現象の解明を目指したさまざまな基礎研究に応用可能です。このため、単に医薬シーズとしての応用だけでなく、プローブ分子の設計や応用を視野に入れるとともに、実際にこれらを用いた生物有機化学研究にも取り組んでいます。
研究活動は、主として学内外の研究室との共同研究により進めています。創薬研究は、分子設計と化学合成だけでなく、生物活性評価(生化学・細胞系・動物実験)や物性・構造評価を同時並行に実施する必要があります。総合薬学研究では、研究活動の初期からこうした共同研究のやりとりを経験することで、研究全体の枠組みやプロセスを学ぶことができます。

鏡像空間からの医薬シーズの探索研究
D-アミノ酸からなる鏡像タンパク質をスクリーニング研究に応用
これまでの創薬研究は、自然界に存在する生物活性を示す天然資源を探したり、生体分子・有機化合物を出発点とした構造最適化研究により行われることが一般的でした。一方、鏡の中に存在する化合物(天然物や生体分子の鏡像体)は、自然界で入手することが困難であり、創薬研究における未開拓の領域の1つです。
私たちは、化学合成によって鏡像タンパク質(すべての配列がD-アミノ酸からなるタンパク質)を取得し、これを用いたスクリーニングにより、鏡の中に存在する化合物(天然物や天然資源)の生物活性を仮想的に評価するプロセスを確立しました。現在、この技術をさまざまな疾患に関連する標的タンパク質に応用することを目指して、タンパク質の化学合成とこれを利用した生物活性評価系の構築に取り組んでいます。
Design and evaluation of stable cysteine-modified monobody scaffolds for mirror-image synthesis
我々はこれまでに、炎症性ケモカインの一種であるMCP-1に対して結合親和性を示す鏡像型モノボディを見出すことに成功しています。本研究では、この鏡像型モノボディのさらなる機能向上を目指し、モノボディを対象とした構造活性相関研究に取り組みました。モノボディに化学修飾を施した複数の誘導体を機能評価に付した結果、結合親和性を維持しつつ優れた熱安定性を示す誘導体を複数同定しました。さらに、最適化された分子骨格を有する鏡像型モノボディが天然型MCP-1に対して結合親和性を示すとともに、さらなる後期修飾に適用可能であることを明らかにしました。

Generating a mirror-image monobody-targeting MCP-1 via TRAP display and chemical protein synthesis
MCP-1は炎症性ケモカインの一種であり、乾癬や関節リウマチをはじめとした様々な疾患との関わりが指摘されています。本研究では、MCP-1の機能を阻害する鏡像型モノボディの取得を目指し、化学合成した鏡像型MCP-1を用いたスクリーニングを実施しました。TRAPディスプレイと呼ばれる改良型mRNAディスプレイ法を用いることで、鏡像型MCP-1に対して高親和性で結合するモノボディの配列を見出しました。この配列をもとに化学合成した鏡像型モノボディは、細胞実験レベルでMCP-1の機能を効果的に抑制するとともに、マウスを用いた実験において低い免疫原性を示しました。

Chemical synthesis of interleukin-6 for mirror-image screening
インターロイキン-6(IL-6)は、炎症疾患に対する魅力的な創薬標的と考えられています。我々は、IL-6の機能を阻害する鏡像型タンパク質の開発を目指し、標的となるIL-6の鏡像型タンパク質の化学合成に取り組みました。183残基からなるIL-6の全長配列を構築するとともに、既知のリガンドとの結合を確認することにより、鏡像型IL-6が活性構造を有することを確認しました。また、鏡像型IL-6を用いたスクリーニングにより、鏡像型IL-6に結合性を示すVHHを見出しました。

Mirror-image human serum albumin domain III as a tool for analyzing site II-dependent molecular interaction
ヒト血清アルブミン(HSA)はヒトの血中に最も豊富に存在するタンパク質の1つであり、HSAに結合する分子はその血中半減期が大きく向上することが知られています。我々は、アルブミン結合性の鏡像型タンパク質の開発を目指して、スクリーニングに用いる鏡像型HSAの部分構造の化学合成に取り組みました。HSA全長の約3分の1にあたるドメインIII(206残基)を鏡像型タンパク質として合成するとともに、ドメインIII中の薬物結合サイトIIを活用した機能評価により、鏡像型HSAドメインIIIが活性構造を有することを確認しました。

Engineering a low-immunogenic mirror-image VHH against vascular endothelial growth factor
我々は以前、VHH(ナノボディ)の鏡像型タンパク質の化学合成法を確立するとともに、鏡像型VHHが低い免疫原性を示し、安全性の高い医薬素材となり得ることを報告しています。本研究では、実際の創薬標的として血管内皮成長因子(VEGF)に結合性を示す鏡像型VHHの探索に取り組みました。化学合成した鏡像型VEGFを用いたスクリーニングにより鏡像型VEGFに結合するVHHの配列を見出しました。この配列を基に化学合成した鏡像型VHHは、VEGFに対して中程度の親和性で結合するとともに、マウスを用いた実験において低い免疫原性を示しました。

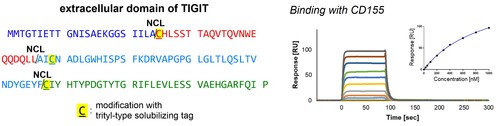
Synthetic studies on the extracellular domain of the T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT) using Trt-K10 solubilizing tags
TIGITはT細胞上に発現する免疫受容体のひとつであり、腫瘍細胞などに発現したCD155と相互作用し、T細胞の活性を抑制する免疫チェックポイントとして機能しています。このため、TIGIT─CD155相互作用の阻害剤は、がん治療などに利用可能であると考えられています。我々は、鏡像型タンパク質を用いた阻害剤探索に利用可能なTIGIT細胞外ドメインの化学合成法を確立しました。
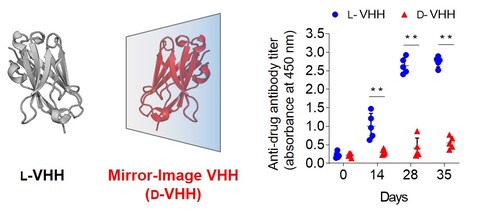
Mirror-image single-domain antibody for a novel nonimmunogenic drug scaffold
抗体医薬などのタンパク質製剤の臨床応用や開発過程では、薬効の減弱や副作用を誘発する抗薬物抗体(投与したタンパク質製剤に対する抗体)の産生を考慮する必要があります。我々は、抗体の結合能を維持する最小ドメインであるVHH抗体の鏡像体(鏡像VHH抗体)の効率的な化学合成法を確立し、マウスへの免疫試験により、鏡像VHH抗体の投与において抗薬物抗体の産生がほとんど誘発されないことを実証しました。鏡像VHH抗体は、抗薬物抗体の産生リスクを低減した新たな医薬品モダリティとして期待されることから、現在、複数の標的に対する鏡像VHH抗体の探索研究を実施しています。
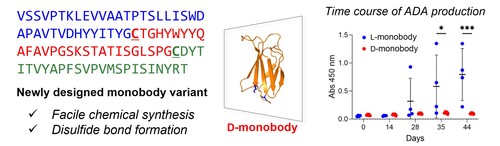
Design and synthesis of monobody variants with low immunogenicity
モノボディは、ヒトフィブロネクチン由来の頑強なドメイン構造に対して可変領域を導入することにより開発された、抗体のような分子認識能を有する人工の小型タンパク質です。我々は、モノボディの鏡像型タンパク質が生体内安定性に優れ免疫原性を低減した有望な医薬材料になると考え、モノボディの化学合成法を確立しました。配列中に導入された2つのCysを足がかりとして簡便に合成されたモノボディの鏡像型タンパク質は、天然型タンパク質と比較して免疫原性が低いことを確認しました。
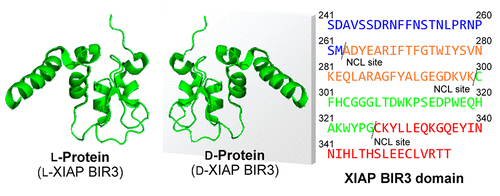
Development of mirror-image screening systems for XIAP BIR3 domain inhibitors
BIRドメインは、カスパーゼの活性を阻害する約70残基からなるZnフィンガー様の構造です。このうち、XIAPタンパク質中のBIR3ドメインに対する阻害剤は、腫瘍細胞をアポトーシスに誘導する薬剤として期待されています。我々は、天然物の鏡像体ライブラリーからXIAP BIR3ドメイン阻害剤の探索を行うために、XIAP BIR3ドメインの化学合成法を確立しました。本手法はBIRドメインのはじめての化学合成例であり、さまざまなBIRドメイン含有タンパク質に対する阻害剤の探索への応用が期待されます。
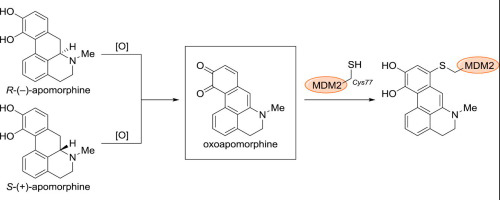
Investigation of the inhibitory mechanism of apomorphine against MDM2-p53 interaction
アポモルヒネは、パーキンソン病の治療薬として用いられている非選択的ドパミン受容体アゴニストです。我々は、化学合成タンパク質を用いた抗がん剤の候補化合物の探索研究において、アポモルヒネの両エナンチオマーがMDM2に作用することを見出しました。また、本来分子認識が異なるはずの2つのエナンチオマーがいずれも生物活性を示すのは、アポモルヒネの酸化によって得られるオキソアポモルヒネがMDM2のCys77のチオール基と共有結合し、その機能を失活させることによるものであることを明らかにしました。
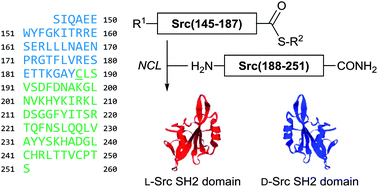
Synthesis of the Src SH2 domain and its application in bioassays for mirror-image screening
Src SH2ドメインは、細胞増殖のシグナル伝達系に関わるアダプタータンパク質です。我々は、天然物の鏡像体化合物群からの医薬品探索を可能にする生物活性評価系の構築の一環で、Src SH2ドメインの天然型タンパク質と鏡像体タンパク質の化学合成法を確立しました。得られたタンパク質は、リン酸化チロシン含有ペプチドとの強力な結合親和性を有しており、複数の生物活性評価系に応用可能であることを明らかにしました。
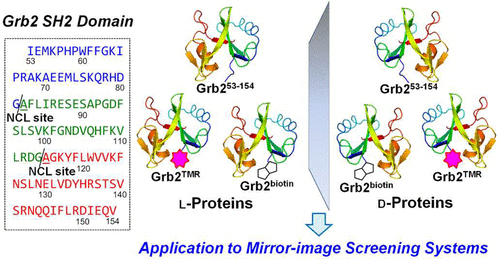
Synthesis of Grb2 SH2 domain proteins for mirror-image screening systems
我々は、以前の研究で、D-タンパク質を用いたスクリーニングにより天然物の鏡像体化合物群からの医薬品探索を実現するプロセスを確立しました。この天然物の鏡像スクリーニングを様々な標的タンパク質に適応させることを目的として、これまでに化学合成例のないSH2ドメインタンパク質の合成研究を行いました。2セグメント及び3セグメントによるGrb2 SH2ドメインの合成法を確立し、蛍光標識基等を有する鏡像体タンパク質を合成しました。また、これらを用いた生物活性評価系を確立し、SH2ドメインタンパク質が鏡像スクリーニングに応用できることを明らかにしました。
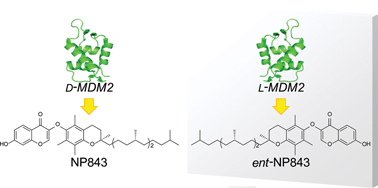
Screening of a virtual mirror-image library of natural products
キラルな天然物の鏡像体は、天然物と同等の化合物特性を有していることから医薬品探索の新たなリソースとして期待されます。我々は、化学合成により調製した鏡像体タンパク質(D-タンパク質)をスクリーニングに用いることで、鏡写しの関係で生体内タンパク質(L-タンパク質)と天然物の鏡像体群との相互作用を効率的に評価する新規探索手法を開発しました。標的分子としてMDM2を選択し上記概念に基づく探索プロセスを実施した結果、ent-NP843をMDM2-p53間の新規阻害剤として見出しました。
ペプチド性天然物からの創薬研究
細胞内で機能するペプチド性二次代謝産物の合成と高活性誘導体の創製
微生物が二次代謝産物として生産するペプチドには、免疫抑制剤であるシクロスポリンに代表されるように、細胞膜を透過して細胞内の標的分子に作用するものがあります。私たちは、微量で強力な生物活性を示すペプチド性天然物について、その化学合成を通して化学構造の決定を試みたり、医薬品としての応用を目指した構造活性相関研究に取り組んでいます。化学合成により得られた天然物やプローブは、その標的分子を決定する生物有機化学研究に応用しています。
Structure and synthesis of amatyemides A and B, cyclic octadepsipeptides from South African stromatolites
Amatyemide A及びBは、南アフリカで採取されたストロマトライトの研究の過程で見出され環状オクタデプシペプチドです。Amatyemide A及びBには、側鎖に長鎖アルキル基を有するα-ヒドロキシ酸や非天然アミノ酸が含まれており、スペクトル解析などの手法で構造決定を行うことが困難でした。我々は、amatyemide A及びBの化学合成プロセスを確立し、天然品と化学合成品の比較によりamatyhemide類の構成要素の立体配置を決定しました。(オレゴン州立大学との共同研究)

Design of synthetic surrogates for the macrolactone linker motif in coibamide A
Coibamide Aは、細胞内でのタンパク質の輸送に関わるトランスロコンSec61に作用してその機能を阻害する環状デプシペプチドです。我々は、coibamide Aの大環状構造のマクロラクトン部分をアルケンやアミドに変換した誘導体を設計・合成し、構造活性相関研究を行いました。これらの誘導体はいずれも優れた生物活性を示し、coibamide AのMeThr5のβ-メチル基及びD-MeAla11のα-メチル基はいずれも生物活性の発現に重要な役割を果たしていることを明らかにしました。(オレゴン州立大学との共同研究)

Design of coibamide A mimetics with improved cellular bioactivity
海洋シアノバクテリア由来のcoibamide Aは、細胞内でのタンパク質の輸送に関わるトランスロコンSec61に作用してその機能を阻害する環状ペプチドです。我々は、coibamide AのMeThr5-D-MeAla11部分をMeLysに置換した誘導体の簡便な化学合成法を確立し、構成アミノ酸の構造活性相関研究を迅速に行うことを可能にしました。また、この手法を用いてTyr(Me)10の構造最適化を試み、biphenylylalanine (Bph)に置換した誘導体がcoibamide Aよりも強力な生物活性を示すことを明らかにしました。(オレゴン州立大学との共同研究)

Coibamide A targets Sec61 to prevent biogenesis of secretory and membrane proteins
Coibamide Aは、強力な細胞増殖抑制活性を示す環状ペプチドで、これまでの抗がん剤にないユニークな生物活性プロファイルを示すことが知られています。我々は、coibamide Aの標的分子を同定する目的で、光親和性官能基を有するcoibamide A誘導体を設計・合成しました。これをプローブとして利用した実験により、coibamide Aの標的分子が小胞体膜上にあるタンパク質輸送体Sec61であることを明らかにしました。(ヘルシンキ大学・オレゴン州立大学との共同研究)

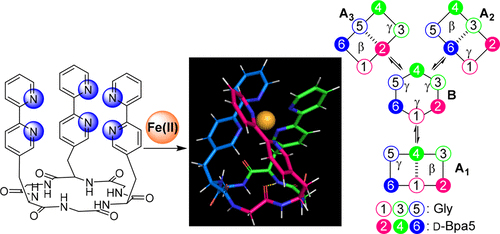
Use of a compact tripodal tris(bipyridine) ligand to stabilize a single metal-centered chirality: stereoselective coordination of iron(II) and ruthenium(II) on a semi-rigid hexapeptide macrocycle
Fe(II)イオンは、非対称型リガンドと錯体を形成すると、Fe(II)中心のキラリティー(Λ型とΔ型)を生じます。我々は、ビピリジン構造を含むα-アミノ酸を3残基含む環状ヘキサペプチドを合成しました。この環状ペプチドはFe(II)と1:1の錯体を形成し、Λ体の金属中心不斉からなる錯体のみが生成しました。各種スペクトル測定および分子動力学シミュレーションにより溶液中のペプチド─Fe(II)錯体の構造を解析したところ、環状ペプチド部分は複数のコンフォメーション間で相互に変化するフレキシブルな状態で存在する一方で、3つのビピリジン部分の相対的な空間配置はほとんど変化せず安定な立体配置の維持に寄与していることが示唆されました。
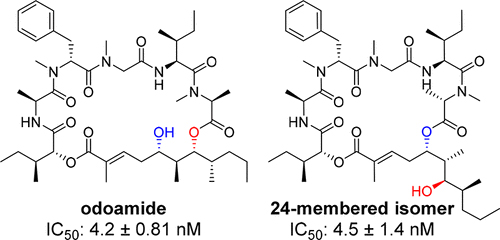
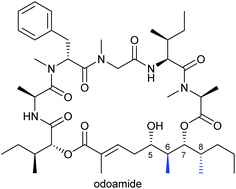
SAR study on odoamide: insights into the bioactivities of aurilide-family hybrid peptide-polyketides
Odoamideは沖縄近海のシアノバクテリア類より単離された強力な細胞増殖抑制活性を示す環状デプシペプチドです。我々は、odoamideの活性に寄与する因子を明らかにすることを目的として、構造活性相関研究に取り組みました。まず、odoamide誘導体の効率的な合成経路を確立するとともに、複数の誘導体を合成し、天然物と同等の活性を有するペプチドを見出しました。また、天然物と24員環異性体が分子内アシル転位反応を介した平衡関係にあり、異性体も天然物と同等の生物活性を有することを明らかにしました。
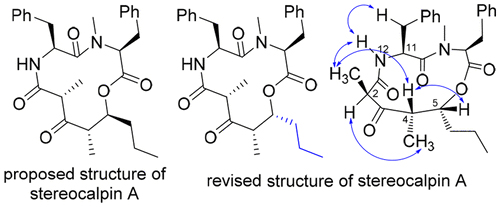
Total synthesis and stereochemical revision of stereocalpin A: mirror-image approach for stereochemical assignments of the peptide-polyketide macrocycle
Stereocalpin Aは南極地方の地衣類より単離された細胞増殖抑制活性を示す環状デプシペプチドです。過去のstereocalpin Aの合成研究によりその提唱構造の誤りが指摘されており、現在まで天然物の立体化学は明らかではありませんでした。我々はstereocalpin Aの立体化学を決定することを目的として、合成研究に取り組みました。まず、stereocalpin Aのポリケチド部分の立体化学が異なる8種類の立体異性体を効率よく合成しました。得られた立体異性体と天然物とのNMRスペクトルの比較により、5位の立体化学が提唱構造とは異なる異性体のNMRスペクトルが天然物のものとよい一致を示しました。これにより、stereocalpin Aの全合成及び、構造修正を達成しました。
Fe(II)-complexation of tripodal hexapeptide ligands with three bidentate triazolylpyridines: induction of metal-centred chirality by peptide macrocyclization
我々は、Fe(III)配位性天然ペプチドの構造をもとにして、Fe(II)配位性ヘキサペプチドを設計しました。このうち、トリアゾリルピリジン含有アミノ酸を1残基おきに配置した環状および直鎖ヘキサペプチドが高いFe(II)親和性を示すことを明らかにしました。この2つのペプチドは同一のアミノ酸配列からなるものですが、金属中心の立体化学に着目すると反対のキラリティーを示すという興味深い現象を見出しました。

Total synthesis of odoamide, a novel cyclic depsipeptide from an Okinawan marine cyanobacterium
Odoamideは沖縄近海に生息するシアノバクテリア類より単離された、強力な細胞増殖抑制活性を有する環状デプシペプチドです。我々は、odoamideの絶対立体配置を明らかにすることを目的として、全合成研究を行いました。ポリケチド部分の絶対立体配置を決定した後、各フラグメントを順次縮合し、odoamideの推定構造を合成しました。合成したodoamideの各種スペクトルデータは天然物とよい一致を示し、odoamideの初の全合成を達成しました。
BACE1阻害剤に関する研究
アルツハイマー病治療薬をめざしたβアミロイド産生と機能調節に関する研究
アルツハイマー病の発症にはβアミロイドの凝集が大きくかかわっています。したがって、このβアミロイドの産生あるいは凝集を止めることができる化合物は極めて有望なアルツハイマー治療薬となります。私たちの研究室では、βアミロイドの産生に必須のBACE1とよばれるプロテアーゼの大量発現に成功し、このプロテアーゼを利用した阻害剤の設計と評価を行っています。私たちはこれまでに、基質配列をもとにしたペプチド性BACE1阻害剤の開発に成功しており、現在この阻害剤の構造情報に基づいた環状阻害剤の合成研究を進めています。また、ヒドロキシプロリンを基盤とした全く新しい骨格を有する低分子阻害剤の開発研究も並行して進めています。
Macrocyclic BACE1 inhibitors with hydrophobic cross-linked structures: Optimization of ring size and ring structure
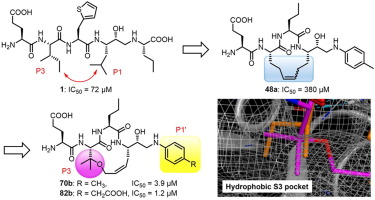
我々は、本研究室で見出した線形BACE1阻害剤をリード化合物として、阻害活性や物性の向上を目指し、P1-P3側鎖間への架橋構造の導入を行いました。まず、最適環サイズの探索として、12~15員環を有する大環状阻害剤を合成・評価したところ、13員環が最も良い活性を示しました。しかし、その阻害活性はリード化合物より大きく低下してしまったため、ドッキングシミュレーションの結果を基に最適環構造の探索を行いました。その結果、P3-β位にジメチル基を導入した70bが、リード化合物を上回る活性を示すことを見出しました。また、得られた環状阻害剤のP1’位にフェニル酢酸を導入することで更なる活性向上が認められることを明らかにしました。
Structure–activity relationship study of hydroxyethylamine isostere and P1′ site structure of peptide mimetic BACE1 inhibitors
我々は、高親和性基質配列とヒドロキシエチルアミン(HEA)型ペプチドイソスターを組み合わせて設計したBACE1阻害剤をリード化合物として、更なる活性向上を目指し、構造活性相関研究に取り組みました。まず、分岐的合成法によりHEA構造を立体選択的に作り分け、R配置を取る無置換HEA構造が活性に最も寄与することを見出しました。また、P1’位の構造について種々検討を行い、芳香環上のパラ位にカルボキシメチル基を有する40gが、既存の阻害薬であるlanabecestatよりも高い活性を有することを明らかにしました。
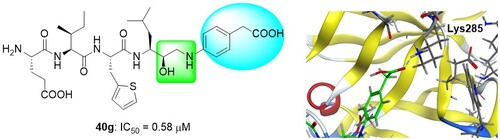
Evaluation of transition-state mimics in a superior BACE1 cleavage sequence as peptide-mimetic BACE1 inhibitors
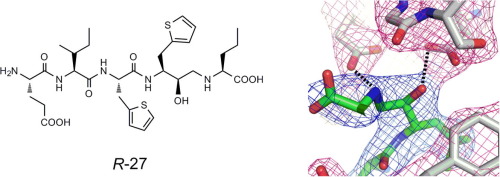
我々は、高親和性基質配列にヒドロキシメチルカルボニル(HMC)イソスター、及びヒドロキシエチルアミン(HEA)イソスターを組み合わせることで新規ペプチド性BACE1阻害剤を設計し、その構造活性相関研究に取り組みました。その結果、イソスター部の水酸基の立体が活性に大きく寄与していること、活性発現に必要な立体はP1位側鎖に対して、HMC型阻害剤ではanti配置、HEA型阻害剤ではsyn配置であることを明らかにしました。
抗ウイルス剤の創製研究
病原性ウイルス由来のタンパク質の構造をもとにしたペプチド・ペプチドミメティックスの設計と合成
病原性ウイルスが引き起こす感染症には、急性期の激しい症状を引き起こすものだけでなく、長期間をかけて重篤な病態へと変化するものがあり、それぞれのウイルスの特性にあわせて薬効を示す優れた治療薬の開発が求められています。 私たちは、新しい抗ウイルス剤の創製に向けた取り組みとして、病原性ウイルスが持つタンパク質の構造や特性にもとづき、その機能や活性を調節・阻害する化合物の分子設計・合成を行っています。
Elucidation of postfusion structures of the measles virus F protein for the structure-based design of fusion inhibitors
我々は、宿主細胞へ感染する際に機能する麻疹ウイルスのFタンパク質の膜融合プロセスに着目し、Fタンパク質が形成するαヘリックス構造からなるタンパク質複合体の構造をX線結晶構造解析により明らかにしました。また、この構造解析の結果をもとにαヘリックス構造の安定性を向上し、標的タンパク質との相互作用を最適化した新規抗ウイルス活性ペプチドを設計・合成しました。(東北大学との共同研究)

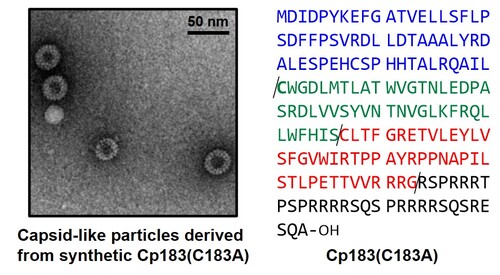
Synthesis of the full-length hepatitis B virus core protein and its capsid formation
B型肝炎ウイルス(HBV)は、肝臓に感染して炎症を引き起こすウイルスであり、持続感染した場合、肝硬変や肝がんへと進展する可能性があります。HBVのウイルスDNAを包み込むカプシドを構成するコアタンパク質は、HBV複製サイクルを阻害するための標的の一つとして注目を集めています。我々は、化合物の鏡像体ライブラリーからのHBVカプシド形成阻害剤の探索を行うために、HBVカプシドの全長コアタンパク質(Cp183)の化学合成法を確立しました。本手法は化学合成によってHBVカプシド様粒子へと導いたはじめての例であり、鏡の中の分子を利用した新規HBV阻害剤の探索への応用が期待されます。
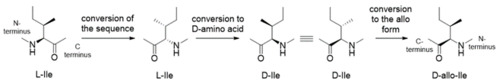
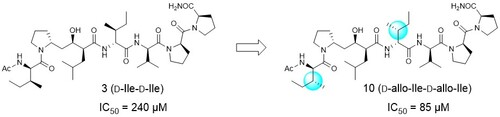
The effects of side-chain configurations of a retro–inverso-type inhibitor on the human T-cell leukemia virus (HTLV)-1 protease
これまでに報告したRI(retro inverso)型HTLV-1プロテアーゼ阻害剤では、基質配列中のL-IleをD-Ileに変換して設計を行っていましたが、本研究ではD-Ileをallo-D-Ileへと置換した誘導体を合成し、阻害活性評価を行いました。その結果、2つのD-Ileをそれぞれallo-D-Ileへと置換することで阻害活性が約3倍向上することを明らかにしました。この結果は、主鎖のみならず側鎖も含めた立体化学の模倣が、RI型阻害剤の設計において重要であることを示しています。
Evaluation of an octahydroisochromene scaffold used as a novel SARS 3CL protease inhibitor
我々は、これまでに報告したSARS 3CLプロテアーゼ阻害剤にオクタヒドロイソクロメン骨格を導入した新規SARS 3CLプロテアーゼ阻害剤を設計し、その阻害能の評価を行いました。その結果、オクタヒドロイソクロメン骨格が想定通りS2ポケットにおける疎水性コアとして有用であること、1位側鎖の置換基は酵素表面と相互作用をしていること、オクタヒドロイソクロメン骨格の絶対立体配置が阻害活性に大きく寄与することを明らかにしました。
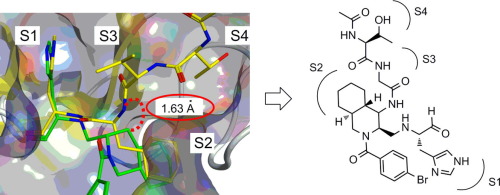
Evaluation of a non-prime site substituent and warheads combined with a decahydroisoquinolin scaffold as a SARS 3CL protease inhibitor
これまでに報告したデカヒドロイソキノリン骨格を有するSARS 3CLプロテアーゼ阻害剤は、S3,S4ポケットにおける相互作用部位が欠落していたことから、その阻害活性は中程度のものでした。そこで我々は、デカヒドロイソキノリン骨格にノンプライム部位に対応する置換基を導入した新規SARS 3CLプロテアーゼ阻害剤を設計し、その阻害能の評価を行いました。その結果、新たに設計した阻害剤は元の阻害剤から2.4倍活性が向上していることを明らかにしました。
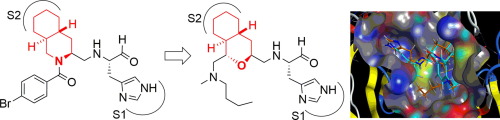
Fused-ring structure of decahydroisoquinolin as a novel scaffold for SARS 3CL protease inhibitors
我々は、基質配列に基づいて設計されたペプチド性SARS 3CLプロテアーゼ阻害剤のP2位側鎖における疎水性相互作用に着目し、側鎖シクロヘキシル基と主鎖α-窒素原子をメチレン鎖で連結したデカヒドロイソキノリン型阻害剤を設計し、その阻害能の評価を行いました。合成した誘導体はいずれも弱いながらも明らかな阻害活性を示したことから、デカヒドロイソキノリン骨格がSARS 3CLプロテアーゼ阻害剤のための新規骨格として有用であることが示されました。またX線結晶構造解析から、(3S,4aR,8aS)の立体を有する骨格がより高活性を示すことを明らかにしました。
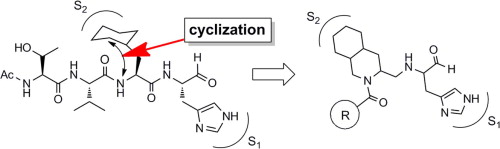
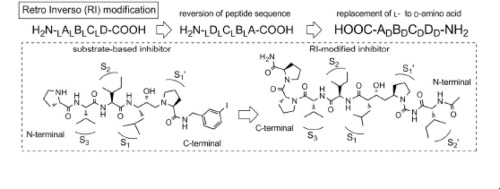
Effect of prime-site sequence of retro-inverso-modified HTLV-1 protease inhibitor
RI(retro inverso)型変換とは、ペプチドにおいて配列の方向と各アミノ酸のキラリティーの両方を逆転させる変換のことであり、生理活性を維持したまま生体内安定性を改善することができると期待される変換法の1つです。我々は、基質配列に基づいて設計されたHTLV-1プロテアーゼ阻害剤にRI型変換を行ったRI型阻害剤をリード化合物として、P2’部位の置換基に関する構造活性相関研究に取り組みました。その結果、D-Ileを導入した誘導体において僅かではありますが活性が向上することを明らかにしました。